Reproducible Analysis & Models for Predicting Genomics Workflow Execution Time
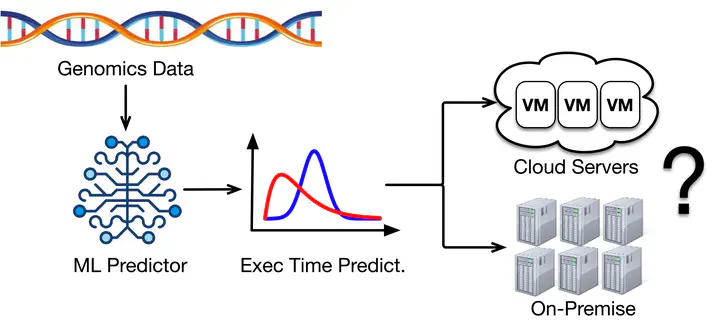
A high-throughput workflow execution system is needed to continuously gain insights from th e increasingly abundant genomics data. However, genomics workflows often have long execution times (e.g., hours to days) due to their large input files. This characteristic presents many complexities when managing systems for genomics workflow execution. Furthermore, based on our observation of a large-scale genomics data processing platform, ~2% of genomics workflows exhibit a tail behavior which multiplied their execution time up to 15x of the median, resulting in weeks of execution.
On the other hand, input files for genomic workflows often vary in quality due to differences in how they are collected. Prior works suggested that these quality differences can affect genomics workflow execution time. Yet, to the best of our knowledge, input quality has never been accounted for in the design of a high-throughput workflow execution system. Even worse, there does not appear to be a consensus on what constitutes ‘input quality,’ at least from a computer systems perspective.
In this project, we seek to analyze a huge dataset from a large-scale genomics processing platform in order to gain insights on how ‘input quality’ affects genomic workflows’ execution times. Following that, we will build machine learning (ML) models for predicting workflow execution time, in particular those which exhibit tail behavior. We believe these insights and models can become the foundation for designing a novel tail-resilient genomics workflow execution system. Along the way, we will ensure that each step of our analysis is reproducible (e.g., in the form of Jupyter notebooks) and make all our ML models open-source (e.g., in the form of pre-trained models). We sincerely hope our work can offload some burdens commonly faced by operators of systems for genomics and, at the same time, benefit future researchers who work on the intersection of computer systems and genomics.
Analyze genomics data quality & build exec. time prediction models
- Topics: genomics, data analysis, machine learning
- Skills: Linux, Python, Matplotlib, Pandas/Numpy, any ML library
- Difficulty: Medium
- Size: 350 hours
- Mentor(s): In Kee Kim
- Contributor(s): Charis Christopher Hulu
Analyze a large-scale trace of genomics workflow execution along with metrics from various genomics alignment tools (e.g., FastQC, Picard, and GATK metrics) and find features that correlate the most with workflow execution time and its tail behavior. Then, based on the results, we will build ML models that accurately predict genomic workflows’ execution times.
Specific tasks:
- Acquire basic understanding of genomics data processing & workflow execution (will be guided by the mentor)
- Reproduce past analysis & models built by prior members of the project
- Propose features from FastQC/Picard/GATK metrics that can be used as a predictor for execution time and tail behavior
- Write a brief analysis as to why those features might work
- Build ML models for predicting execution time
- Package the analysis in the form of Jupyter notebooks
- Package the models in a reloadable format (e.g., pickle)
In Kee Kim
Associate Professor, University of Georgia
In Kee Kim is currently an Associate Professor in the School of Computing at the University of Georgia. He obtained his Ph.D. in Computer Science from the University of Virginia in 2018.